Compositional Perturbation Autoencoder (CPA)


Project description
CPA - Compositional Perturbation Autoencoder



What is CPA?
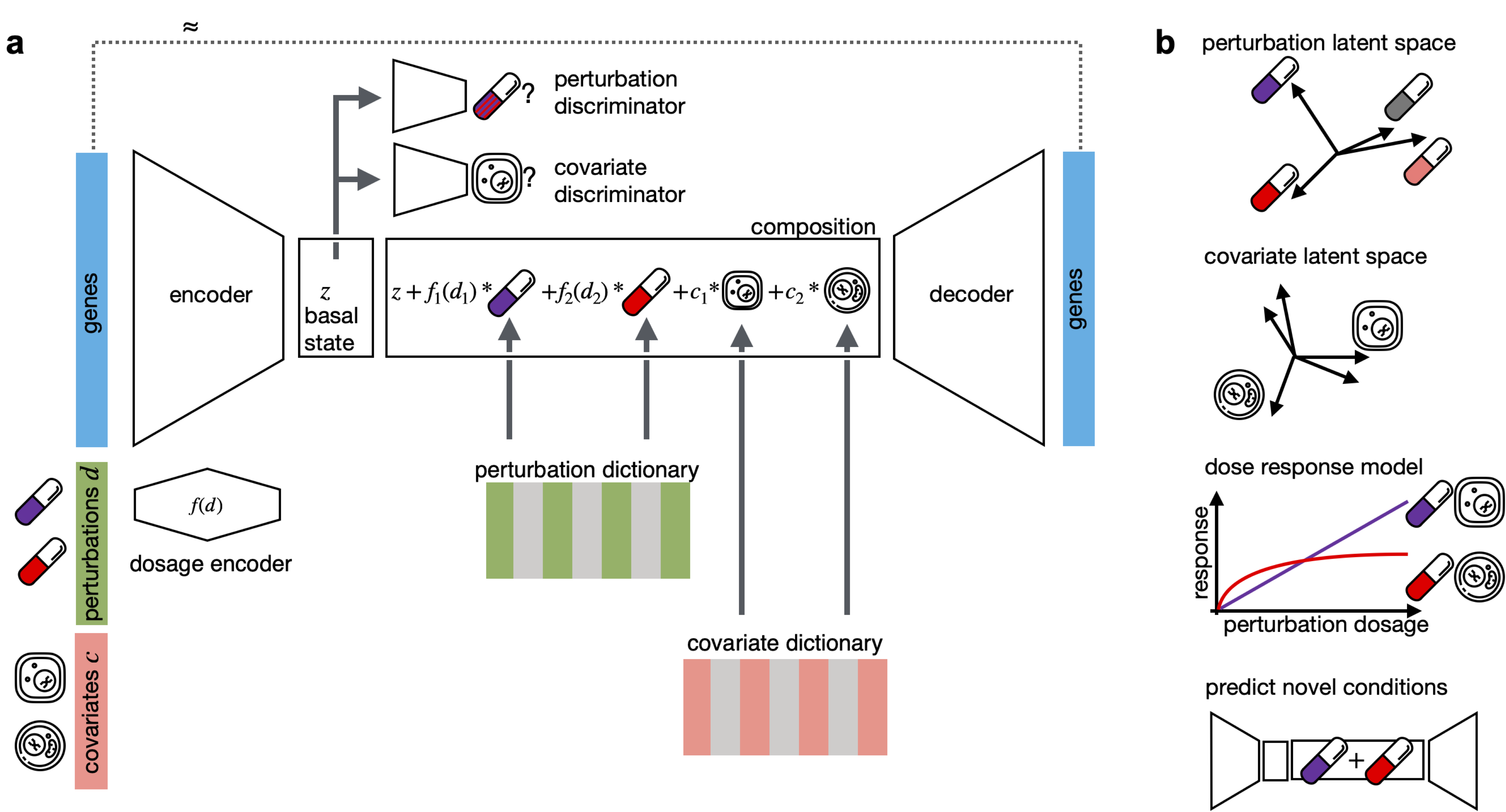
CPA is a framework to learn the effects of perturbations at the single-cell level. CPA encodes and learns phenotypic drug responses across different cell types, doses, and combinations. CPA allows:
- Out-of-distribution predictions of unseen drug and gene combinations at various doses and among different cell types.
- Learn interpretable drug and cell-type latent spaces.
- Estimate the dose-response curve for each perturbation and their combinations.
- Transfer pertubration effects from on cell-type to an unseen cell-type.
- Enable batch effect removal on a latent space and also gene expression space.
Installation
Installing CPA
You can install CPA using pip and also directly from the github to access latest development version. See detailed instructions here.
How to use CPA
Several tutorials are available here to get you started with CPA. The following table contains the list of tutorials:
| Description | Link |
|---|---|
| Predicting combinatorial drug perturbations |  - -  |
| Predicting unseen perturbations uisng external embeddings enabling the model to predict unseen reponses to unseen drugs | - |
| Predicting combinatorial CRISPR perturbations | - |
| Context transfer (i.e. predict the effect of a perturbation (e.g. disease) on unseen cell types or transfer perturbation effects from one context to another) demo on IFN-β scRNA perturbation dataset | - |
| Batch effect removal in gene expression and latent space | - |
How to optmize CPA hyperparamters for your data
We provide an example script to use the built-in hyperparameter optimization function in CPA (based on scvi-tools hyperparam optimizer). You can find the script at examples/tune_script.py.
After the hyperparameter optimization using tune_script.py is done, result_grid.pkl is saved in your current directory using the pickle library. You can load the results using the following code:
import pickle
with open('result_grid.pkl', 'rb') as f:
result_grid = pickle.load(f)
From here, you can follow the instructions in the Ray Documentations to analyze the run, and choose the best hyperparameters for your data.
You can also use the integration with wandb to log the hyperparameter optimization results. You can find the script at examples/tune_script_wandb.py. --> use_wandb=True
Everything is based on Ray Tune. You can find more information about the hyperparameter optimization in the Ray Tune Documentations.
The tuner is adapted and adjusted from scvi-tools v1.2.0 (unreleased) release notes
Datasets and Pre-trained models
Datasets and pre-trained models are available here.
Recepie for Pre-processing a custom scRNAseq perturbation dataset
If you have access to you raw data, you can do the following steps to pre-process your dataset. A raw dataset should be a scanpy object containing raw counts and available required metadata (i.e. perturbation, dosage, etc.).
Pre-processing steps
-
Check for required information in cell metadata: a) Perturbation information should be in
adata.obs. b) Dosage information should be inadata.obs. In cases like CRISPR gene knockouts, disease states, time perturbations, etc, you can create & add a dummy dosage in youradata.obs. For example:adata.obs['dosage'] = adata.obs['perturbation'].astype(str).apply(lambda x: '+'.join(['1.0' for _ in x.split('+')])).values
c) [If available] Cell type information should be in
adata.obs. d) [Multi-batch integration] Batch information should be inadata.obs. -
Filter out cells with low number of counts (
sc.pp.filter_cells). For example:sc.pp.filter_cells(adata, min_counts=100)
[optional]
sc.pp.filter_genes(adata, min_counts=5)
-
Save the raw counts in
adata.layers['counts'].adata.layers['counts'] = adata.X.copy()
-
Normalize the counts (
sc.pp.normalize_total).sc.pp.normalize_total(adata, target_sum=1e4, exclude_highly_expressed=True)
-
Log transform the normalized counts (
sc.pp.log1p).sc.pp.log1p(adata)
-
Highly variable genes selection: There are two options: 1. Use the
sc.pp.highly_variable_genesfunction to select highly variable genes.python sc.pp.highly_variable_genes(adata, n_top_genes=5000, subset=True)2. (Highly Recommended specially for Multi-batch integration scenarios) Use scIB's highly variable genes selection function to select highly variable genes. This function is more robust to batch effects and can be used to select highly variable genes across multiple datasets.python import scIB adata_hvg = scIB.pp.hvg_batch(adata, batch_key='batch', n_top_genes=5000, copy=True)
Congrats! Now you're dataset is ready to be used with CPA. Don't forget to save your pre-processed dataset using adata.write_h5ad function.
Support and contribute
If you have a question or new architecture or a model that could be integrated into our pipeline, you can post an issue
Reference
If CPA is helpful in your research, please consider citing the Lotfollahi et al. 2023
@article{lotfollahi2023predicting,
title={Predicting cellular responses to complex perturbations in high-throughput screens},
author={Lotfollahi, Mohammad and Klimovskaia Susmelj, Anna and De Donno, Carlo and Hetzel, Leon and Ji, Yuge and Ibarra, Ignacio L and Srivatsan, Sanjay R and Naghipourfar, Mohsen and Daza, Riza M and
Martin, Beth and others},
journal={Molecular Systems Biology},
pages={e11517},
year={2023}
}
Release history Release notifications | RSS feed


Download files
Download the file for your platform. If you're not sure which to choose, learn more about installing packages.
Source Distribution
Built Distribution
File details
Details for the file cpa_tools-0.8.8.tar.gz.
File metadata
- Download URL: cpa_tools-0.8.8.tar.gz
- Upload date:
- Size: 51.8 kB
- Tags: Source
- Uploaded using Trusted Publishing? No
- Uploaded via: poetry/1.6.1 CPython/3.9.18 Linux/6.5.0-44-generic
File hashes
| Algorithm | Hash digest | |
|---|---|---|
| SHA256 | 7c30086ec23133bf338aef8d290c234cec5ace42480743af3c5d6f6d65235474 |
|
| MD5 | f4a9dec7850a41ae3ce6186c31f47033 |
|
| BLAKE2b-256 | 7cbfa85a0a3844e5c59bb7fd36e0afa39c9d0d2cbdb8cbb3bb00f847e0d29740 |
File details
Details for the file cpa_tools-0.8.8-py3-none-any.whl.
File metadata
- Download URL: cpa_tools-0.8.8-py3-none-any.whl
- Upload date:
- Size: 51.9 kB
- Tags: Python 3
- Uploaded using Trusted Publishing? No
- Uploaded via: poetry/1.6.1 CPython/3.9.18 Linux/6.5.0-44-generic
File hashes
| Algorithm | Hash digest | |
|---|---|---|
| SHA256 | 8d913045cb3757299757a91068e0885e93db9ae17acc2185aae2ecef2ae7525d |
|
| MD5 | f6afed0682bb8ceb64c98f6c75e75382 |
|
| BLAKE2b-256 | 6828c5a4d5c575861c1872edfc938026044af8f8330b6308950ffa86e1df745e |







