Python package to lazily generate synthetic genomic sequences for training od Keras models.

Project description







Python package to lazily generate synthetic genomic sequences for training of Keras models.
How do I install this package?
As usual, just download it using pip:
pip install keras_synthetic_genome_sequenceTests Coverage
Since some software handling coverages sometime get slightly different results, here’s three of them:



Usage examples
To use GapSequence to train your keras model you will need to obtain statistical metrics for the biological gaps you intend to mimic in your synthetic gaps.
To achieve this, this package offers an utility called get_gaps_statistics, which allows you to obtain the mean and covariance of gaps in a given genomic assembly.
The genomic assembly is automatically downloaded from UCSC using ucsc_genomes_downloader, then the gaps contained within are extracted and their windows is expanded to the given one, after filtering for the given max_gap_size, as you might want to limit the gaps size to a relatively small one (gaps can get in the tens of thousands of nucleotides, for instance in the telomeres).
Let’s start by listing all the important parameters:
assembly = "hg19"
window_size = 200
batch_size = 128Now we can start by retrieving the gaps statistics:
from keras_synthetic_genome_sequence.utils import get_gaps_statistics
number, mean, covariance = get_gaps_statistics(
assembly=assembly,
max_gap_size=100,
window_size=window_size
)
print("I have identified {number} gaps!".format(number=number))Now you must choose the ground truth on which to apply the synthetic gaps, for instance the regions without gaps in the genomic assembly hg19, chromosome chr1. These regions will have to be tasselized into smaller chunks that are compatible with the shape you have chosen for the gap statistics window_size. We can retrieve these regions as follows:
from ucsc_genomes_downloader import Genome
from ucsc_genomes_downloader.utils import tessellate_bed
genome = Genome(assembly, chromosomes=["chr1"])
ground_truth = tessellate_bed(genome.filled(), window_size=window_size)The obtained pandas DataFrame will have a bed-like format and look as follows:
chrom |
chromStart |
chromEnd |
|
|---|---|---|---|
0 |
chr1 |
10000 |
10200 |
1 |
chr1 |
10200 |
10400 |
2 |
chr1 |
10400 |
10600 |
3 |
chr1 |
10600 |
10800 |
4 |
chr1 |
10800 |
11000 |
Now we are ready to actually create the GapSequence:
from keras_synthetic_genome_sequence import GapSequence
gap_sequence = GapSequence(
assembly=assembly,
bed=ground_truth,
gaps_mean=mean,
gaps_covariance=covariance,
batch_size=batch_size
)Now, having a model that receives as input and output shape (batch_size, window_size, 4), we can train it as follows:
model = build_my_denoiser()
model.fit_generator(
gap_sequence,
steps_per_epoch=gap_sequence.steps_per_epoch,
epochs=2,
shuffle=True
)Happy denoising!
Comparison between biological and synthetic distributions
The following images refer to the biological and synthetic distributions of gaps in the hg19, hg38, mm9 and mm10 genomic assembly, considering gaps with length to up 100 nucleotides and total window size 1000. The threshold used to convert to integer the multivariate gaussian distribution is 0.4, the default value used within the python package.
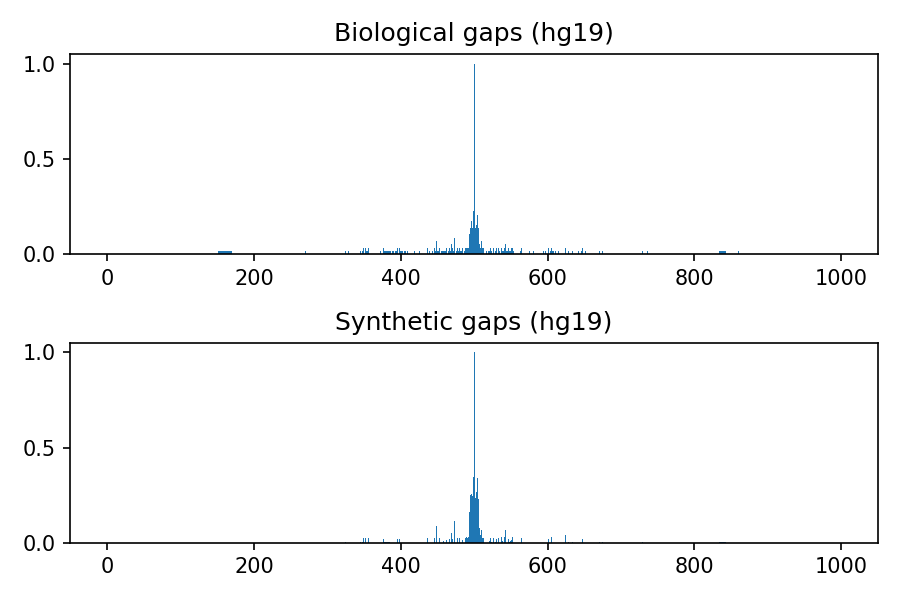
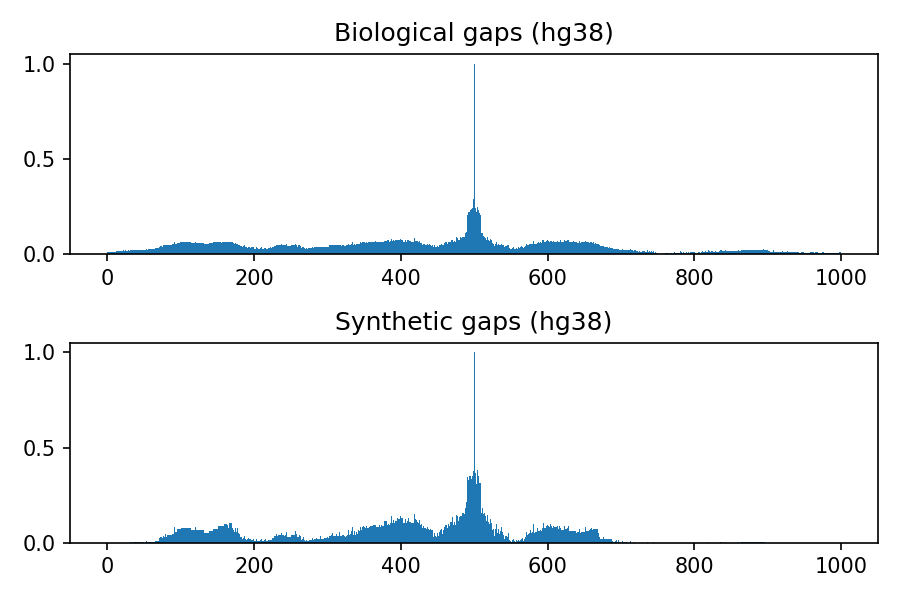
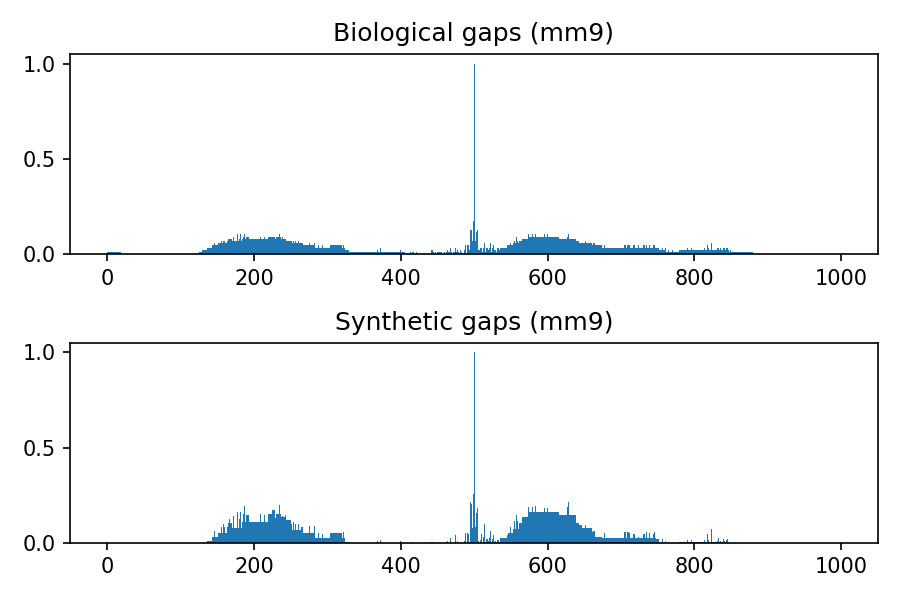
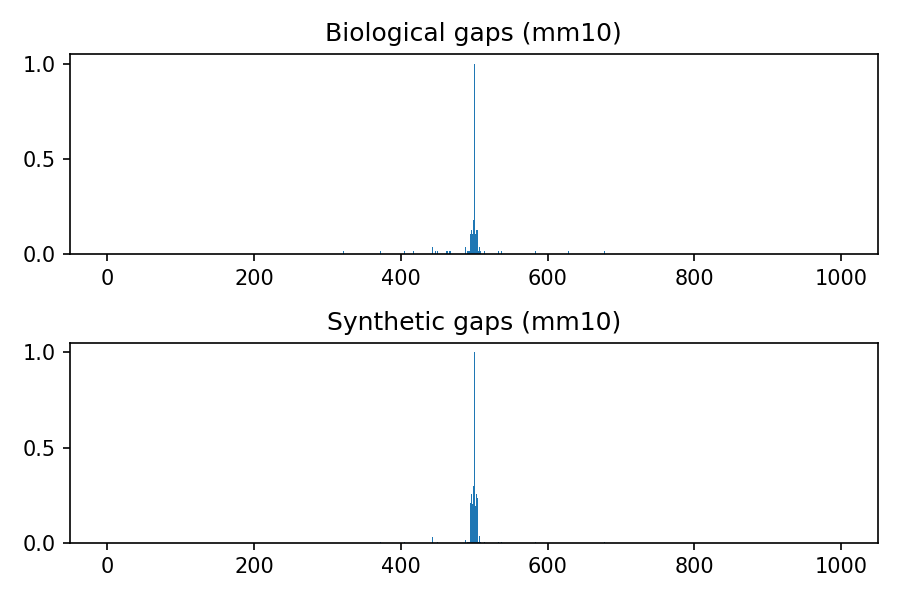
Release history Release notifications | RSS feed


Download files
Download the file for your platform. If you're not sure which to choose, learn more about installing packages.
Source Distribution
Hashes for keras_synthetic_genome_sequence-1.0.7.tar.gz
| Algorithm | Hash digest | |
|---|---|---|
| SHA256 | 39bd5ef35e05ff2f7d332b824b03037de4c1d7b32538e48570e560946c901d21 |
|
| MD5 | 58af9928fc8c32cae80fdab495e50bd9 |
|
| BLAKE2b-256 | e57c701f0b6801b146c6b406b7761322a3ad5344f0404ee5f545a01bebd01383 |







