scvelo - stochastic single cell RNA velocity


Project description



scVelo - stochastic single cell RNA velocity
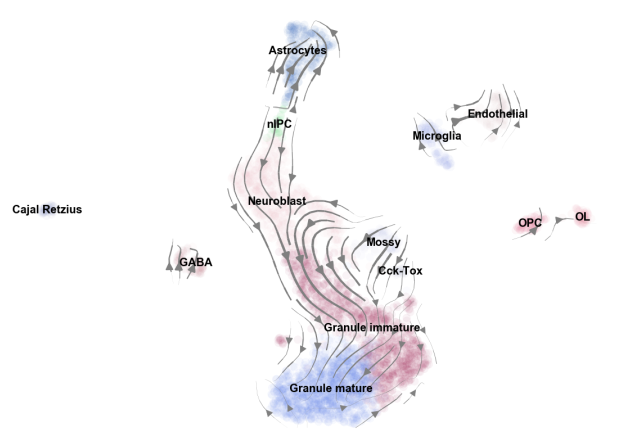
scVelo is a scalable toolkit for estimating and analyzing stochastic RNA velocities in single cells.
The concept of RNA velocity
RNA velocity enables you to infer directionality in your data by superimposing splicing information.
Every cell simultaneously contains newly transcribed (unspliced) pre-mRNA and mature (spliced) mRNA, the former detectable by the presence of introns. La Manno et al., (2018) have shown that the timescale of cellular development is comparable to the kinetics of mRNA lifecycle. Thus, estimating how much mRNA is produced from pre-mRNA, compared to how much of it degrades, enables us to predict its change in the near future. A balance of mRNA production to degradation indicates homeostasis while imbalance indicates a dynamic behaviour, i.e. induction or repression in gene expression. A systematic quantification of the relationship yields RNA velocity, the time derivative of mRNA abundance which tells us how gene expression evolves over time. Aggregating over all genes, we obtain a velocity vector which can be projected into a lower-dimensional embedding to show the movement of the individual cells.
Main features of scVelo
While the potentiality of RNA velocity sounds very promising, several limitations restrict the ability to make truthful predictions. Two of the main challenges, to bear in mind when interpreting results, are the following:
First, key for velocity estimation is to find the steady-state rates where production and degradation are balanced. In the deterministic model that is simply obtained with a linear regression fit on the extreme quantiles (that is where the steady states are most likely located). Thus, a steady-state population is assumed which may be realistic only for genes expressed in populations of terminally differentiated cells.
Second, while velocities can be directly projected into PCA space, that is usually not applicable to non-linear local dimension reduction such as tSNE or UMAP. For these embeddings, probabilities for potential cell transitions are computed, where a high probability corresponds to a high correlation with the velocity vector. The projection is then obtained as expected mean direction given these transition probabilities. Hence, whether the vector field in the embedding truthfully retains the velocities of the high-dimensional space depends on sampling, i.e. whether a cell that reflects the predicted future state well enough is present in the data.
scVelo addresses these challenges as follows:
1) In order to obtain steady-state rates, scVelo uses a stochastic formulation by modeling transcription, splicing and degradation as probabilistic events. It incorporates intrinsic expression variability to better capture the steady states. We will release soon (alongside publishing on biorxiv) a very powerful extension of our model that will not be subject to the steady-state assumption anymore.
2) In order to improve truthfulness of embedded velocities, we incorporate negative correlations (projected into opposite direction) and transition confidence (length of arrows reflect speed of direction but also confidence of transitioning in that direction). For the latter we further established several confidence scores, that encourage to have a critical view on the resulting arrows.
3) There are multiple extensions that can be easily explored, including terminal states (root and end points), pseudotemporal ordering based on velocities, infer directionality in abstracted graph and many more.
scVelo is compatible with scanpy (Wolf et al., 2018). Making use of sparse implementation, iterative neighbors search and other techniques, it is remarkably efficient in terms of memory and runtime without loss in accuracy and runs easily on your local machine (30k cells in a few minutes).
Installation
scVelo requires Python 3.6 or later. We recommend to use Miniconda.
Install scVelo from PyPi using:
pip install -U scvelo
or from source using:
pip install git+https://github.com/theislab/scvelo
Parts of scVelo require (optional):
conda install -c conda-forge numba pytables louvain
The splicing data can be obtained using the velocyto command line interface.
scVelo in action
Import scvelo as:
import scvelo as scv
For beautiful visualization you can change the matplotlib settings to our defaults with:
scv.settings.set_figure_params('scvelo')
Read your data
Read your data file (loom, h5ad, csv, …) using:
adata = scv.read(filename, cache=True)
which stores the data matrix (adata.X), annotation of cells / observations (adata.obs) and genes / variables (adata.var), unstructured annotation such as graphs (adata.uns) and additional data layers where spliced and unspliced counts are stored (adata.layers) .
If you already have an existing preprocessed adata object you can simply merge the spliced/unspliced counts via:
ldata = scv.read(filename.loom, cache=True) adata = scv.utils.merge(adata, ldata)
If you do not have a datasets yet, you can still play around using one of the in-built datasets, e.g.:
adata = scv.datasets.dentategyrus()
Preprocessing
For velocity estimation basic preprocessing (i.e. gene selection and normalization) is sufficient, e.g. using:
scv.pp.filter_and_normalize(adata, **params)
For velocity estimation we need the first- and second-order moments (basically means and variances), computed with:
scv.pp.moments(adata, **params)
Velocity Tools
The core of the software is the efficient and robust estimation of velocities, obtained with:
scv.tl.velocity(adata, mode='stochastic', **params)
The velocities are vectors in gene expression space obtained by solving a stochastic model of transcriptional dynamics. The solution to the deterministic model is obtained by setting mode='deterministic'.
The velocities are stored in adata.layers just like the count matrices.
Now we would like to predict cell transitions that are in accordance with the velocity directions. These are computed using cosine correlation (i.e. find potential cell transitions that correlate with the velocity vector) and are stored in a matrix called velocity graph:
scv.tl.velocity_graph(adata, **params)
Using the graph you can then project the velocities into any embedding (such as UMAP, e.g. obtained with scanpy):
scv.tl.velocity_embedding(adata, basis='umap', **params)
Note, that translation of velocities into a graph is only needed for non-linear embeddings. In PCA space you can skip the velocity graph and directly project into the embedding using scv.tl.velocity_embedding(adata, basis='pca', direct_projection=True).
Visualization
Finally the velocities can be projected and visualized in any embedding (e.g. UMAP) on single cell level, grid level, or as streamplot:
scv.pl.velocity_embedding(adata, basis='umap', **params) scv.pl.velocity_embedding_grid(adata, basis='umap', **params) scv.pl.velocity_embedding_stream(adata, basis='umap', **params)
For every tool module there is a plotting counterpart, which allows you to examine your results in detail, e.g.:
scv.pl.velocity(adata, var_names=['gene_A', 'gene_B'], **params) scv.pl.velocity_graph(adata, **params)
Docs & Feedback
I recommend going through the documentation.
Your feedback, in particular any issue you stumble upon, is highly appreciated and addressed to feedback@scvelo.de.
Release history Release notifications | RSS feed


Download files
Download the file for your platform. If you're not sure which to choose, learn more about installing packages.
Source Distribution
Built Distribution
File details
Details for the file scvelo-0.1.21.tar.gz.
File metadata
- Download URL: scvelo-0.1.21.tar.gz
- Upload date:
- Size: 109.1 kB
- Tags: Source
- Uploaded using Trusted Publishing? No
- Uploaded via: python-requests/2.22.0
File hashes
| Algorithm | Hash digest | |
|---|---|---|
| SHA256 | e026984ac6fd96570840c9ddb73b1caca2f35056c2dd9c56b0f40df7fdbe13a2 |
|
| MD5 | 799444925eaaaf090a879bdadb6df13c |
|
| BLAKE2b-256 | c1956e513608d21e36569826e9200118f8621699cbbeffd1cbc3bdaddc5fe4c4 |
File details
Details for the file scvelo-0.1.21-py3-none-any.whl.
File metadata
- Download URL: scvelo-0.1.21-py3-none-any.whl
- Upload date:
- Size: 391.9 kB
- Tags: Python 3
- Uploaded using Trusted Publishing? No
- Uploaded via: python-requests/2.22.0
File hashes
| Algorithm | Hash digest | |
|---|---|---|
| SHA256 | 0eaf59b5d4ca52415c2af380f3c1c1764236efb955022aa4d76c4197856e056d |
|
| MD5 | 45e29f92cda623ef70adddd9792e0989 |
|
| BLAKE2b-256 | 76faeb7cf52de5529f7983a5b187f9d541a5a10946e96239afa0a251aca4333c |







