A tool for the recovery of unassembled telomeres from soft-clipped read alignments.

Project description

Teloclip
A tool for the recovery of unassembled telomeres from soft-clipped read alignments.
Table of contents
- About Teloclip
- Options and Usage
- Example Usage
- Options
- Citing Teloclip
- Publications using Teloclip
- Issues
- License
About Teloclip
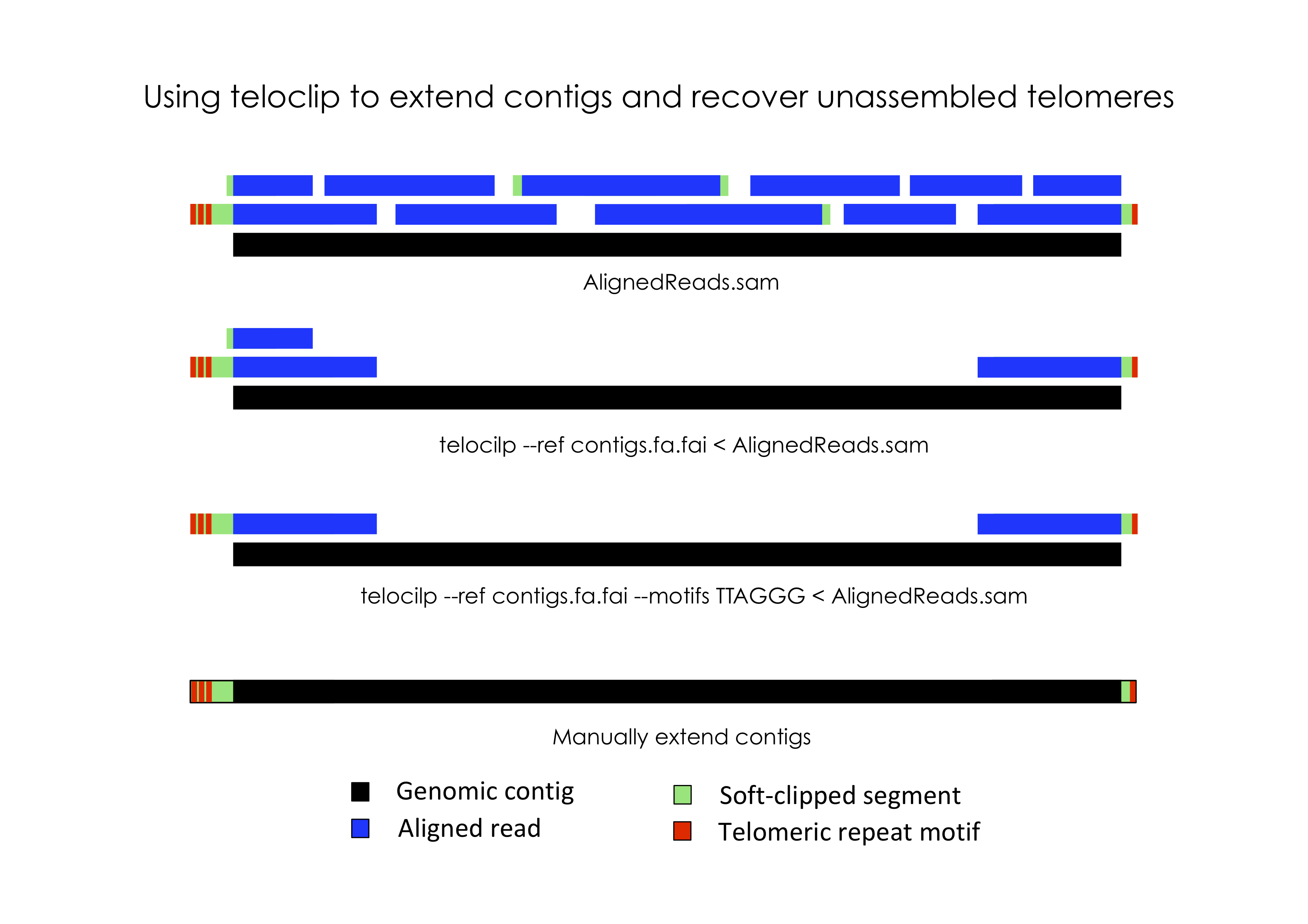
In most eukaryotic species, chromosomes terminate in repetitive telomeric sequences. A complete genome assembly should ideally comprise chromosome-level contigs that possess telomeric repeats at each end. However, genome assemblers frequently fail to recover these repetitive features, instead producing contigs that terminate immediately prior to their location.
Teloclip is designed to recover long-reads that can be used to extend draft contigs and resolve missing telomeres (short-read alignments may also be processed with teloclip). It does this by searching alignments of raw long-read data (i.e. Pacbio or ONT reads mapped with Minimap2) for 'clipped' alignments that occur at the ends of draft contigs. A 'clipped' alignment is produced where the end of a read is not part of its best alignment. This can occur when a read extends past the end of an assembled contig.
Information about segments of a read that were aligned or clipped are stored in SAM formatted alignments as a CIGAR string. Teloclip parses these strings to determine if a read has been clipped at one or both ends of a contig.
Optionally, teloclip can screen overhanging reads for telomere-associated motifs (i.e. 'TTAGGG' / 'CCCTAA') and report only those containing a match.
Teloclip is based on concepts from Torsten Seemann's excellent tool samclip. Samclip can be used to remove clipped alignments from a samfile prior to variant calling.
Options and Usage
Installation
Teloclip requires Python >= 3.8.
There are 4 options available for installing Teloclip locally:
- Install from PyPi.
This or Bioconda will get you the latest stable release.
pip install teloclip
- Install from Bioconda.
conda install -c bioconda teloclip
- Pip install directly from this git repository.
This is the best way to ensure you have the latest development version.
pip install git+https://github.com/Adamtaranto/teloclip.git
- Clone from this repository and install as a local Python package.
Do this if you want to edit the code.
git clone https://github.com/Adamtaranto/teloclip.git && cd teloclip && pip install -e .
Verify installation
# Print version number and exit.
teloclip --version
# > teloclip 0.0.4
# Get usage information
teloclip --help
Run with Gitpod
Alternatively, launch a Gitpod Workspace with teloclip, samtools, and minimap2 pre-installed.
Example Usage
Basic use case:
First index the reference assembly
# Create index of reference fasta
samtools faidx ref.fa
Reading alignments from SAM file
# Read alignment input from sam file and write overhang-reads to stout
teloclip --ref ref.fa.fai in.sam
# Read alignment input from stdin and write stdout to file
teloclip --ref ref.fa.fai < in.sam > out.sam
Reading and writing BAM alignments
BAM files are binary sam files, they contain all the same information but take up much less storage space. You can use bam files with teloclip like this:
# Read alignments from bam file, pipe sam lines to teloclip, sort overhang-read alignments and wite to bam file
samtools view -h in.bam | teloclip --ref ref.fa.fai | samtools sort > out.bam
Streaming SAM records from aligner
# Map PacBio long-reads to ref assembly,
# return alignments clipped at contig ends,
# write to sorted bam.
minimap2 -ax map-pb ref.fa pacbio_reads.fq.gz | teloclip --ref ref.fa.fai | samtools sort > out.bam
# Map reads to reference,
# Exclude non-primary alignments.
# Return alignments clipped at contig ends,
# write to sorted bam.
minimap2 -ax map-pb ref.fa pacbio_reads.fq.gz | samtools view -h -F 0x100 | teloclip --ref ref.fa.fai | samtools sort > out.bam
Report clipped alignments containing target motifs
# Report alignments which are clipped at a contig end
# AND contain >=1 copy of the telomeric repeat "TTAGGG" (or its reverse complement "CCCTAA") in the clipped region.
samtools view -h in.bam | teloclip --ref ref.fa.fai --motifs TTAGGG | samtools sort > out.bam
# Report alignments which are clipped at a contig end
# AND contain >=1 copy of the telomeric repeat "TTAGGG" (or its reverse complement "CCCTAA") ANYWHERE in the read.
samtools view -h in.bam | teloclip --ref ref.fa.fai --motifs TTAGGG --matchAny | samtools sort > out.bam
# To change the minimum number of consecutive repeats required for a match, simply extend the search motif.
# In this example 3 TTAGGG are required for a positive match.
samtools view -h in.bam | teloclip --ref ref.fa.fai --motifs TTAGGGTTAGGGTTAGGG | samtools sort > out.bam
Matching noisy target motifs
Raw long-reads can contain errors in the length of homopolymer tracks. If the --fuzzy option is set, motifs will be converted to regex patterns that allow the number of repeated bases to vary by +/- 1.
i.e. "TTAGGG" -> "T{1,3}AG{2,4}". This pattern will match TTAGG TTAGGGG TAGG TTTAGGG etc.
To reduce off target matching you can increase to minimum required number of motif matches with "--min_repeats".
# Compress homopolymers in query motifs and clipped regions to compensate for errors in raw PacBio or ONP data.
# i.e. The motif 'TTAGGGTTAGGG' becomes 'TAGTAG' and will match 'TTTTTAAAGGTTTAAGGG'.
samtools view -h in.bam | teloclip --ref ref.fa.fai --noPoly --motifs TTAGGGTTAGGG | samtools sort > out.bam
Extract clipped reads
teloclip-extract will write overhanging reads to separate fasta files for each reference contig end. The clipped region of each read is masked as lowercase in output fasta files.
Collections of reads that overhang a contig end can be assembled with miniasm into a single segment before being used to extend the contig. The final telemere-extended assembly should be polished (i.e. with Racon or Pilon) to correct errors in the raw long-read extensions.
# Find clipped alignments containing motif 'TTAGGG' and write reads to separate fasta files for each reference contig end.
samtools view -h in.bam | teloclip --ref ref.fa.fai --motifs TTAGGG | teloclip-extract --refIdx ref.fa.fai --extractReads --extractDir SplitOverhangs
Optional Quality Control
Additional filters
Users may wish to exclude reads below a minimum length or read quality score to reduce the risk of incorrect alignments.
In some cases it may be also be useful to prioritise primary alignments. This can be done by pre-filtering alignments with samtools view. You can decode sam flags here.
# Exclude secondary alignments.
samtools view -h -F 0x100 in.sam | teloclip --ref ref.fa.fai > noSA.sam
Pre-corrected Data
Some assembly tools, such as Canu, preform pre-correction of long-reads through iterative overlapping and correction prior to assembly. Corrected reads are trimmed based on coverage to remove low-confidence ends.
This trimming step can result in loss of distal telomeric sequences and so these reads should NOT be used with Teloclip.
However, long-reads that have been error-corrected using Illumina data with tools such as LoRDEC or HALC should be fine.
Generally speaking, raw long-reads will be fine for extending your contigs. Any errors in the extended region can be corrected with a round of polishing with short-read data using Pilon.
Extending contigs
Before using terminal alignments identified by Teloclip to extend contigs you should inspect the alignments in a genome browser that displays information about clipped reads, such as IGV.
Check for conflicting soft-clipped sequences. These indicate non-specific read alignments. You may need to tighten your alignment criteria or manually remove low-confidence alignments.
After manually extending contigs the revised assembly should be re-polished using available long and short read data to correct indels present in the raw long-reads.
Finally, validate the updated assembly by re-mapping long-read data and checking for alignments that extend into revised contig ends.
Alternative use cases
Illumina data
Teloclip will also work fine with aligned short read data, which has a far lower error rate than single-molecule long-read data.
However, there are obvious limits to the distance that a contig may be extended with shorter reads.
Teloclip does not use information from paired-reads.
Merging existing assemblies
You may have assemblies for your genome generated with different assemblers/configurations or data types (i.e. Illumina, PacBio, ONT) which vary in their success in assembling individual telomeres.
These alternative assemblies can be treated as pseudo-long-reads and aligned to a reference using Minimap2.
Teloclip can identify aligned contigs that can be used to extend those in the reference set.
Be cautious of short contigs that may align to may repetative sub-telomeric regions and result non-specific extension of contigs.
Also beware of low-complexity telomeric regions on different chromosomes aligning to each other and resulting in end-to-end fusions.
# Align alternative assembly contigs to reference and report overhang alignments. Ignore secondary alignments.
minimap2 -ax asm5 ref.fa asm.fa | samtools view -h -F 0x100 | teloclip --ref ref.fa.fai | samtools sort > asm2ref.bam
Circularising Mitochondrial / Bacterial genomes
Using default settings, teloclip will report alignments with clipped regions extending past linear contig ends.
Reads can be extracted from these alignments using circlator's bam2reads and re-aligned to an assembly graph in Bandage to help identify uncircularised contigs.
Options
Teloclip Options
Run teloclip --help to view the programs' most commonly used options:
Usage: teloclip [-h] [--version] --refIdx REFIDX [--minClip MINCLIP] [--maxBreak MAXBREAK]
[--motifs MOTIFS] [--noRev NOREV] [--noPoly NOPOLY] [--matchAny MATCHANY]
[samfile]
Required:
--refIdx REFIDX Path to fai index for reference fasta. Index fasta using `samtools faidx FASTA`
Positional arguments:
samfile Input SAM can be added as the first positional argument after flagged options.
If not set teloclip will read from stdin.
Optional:
--minClip Require clip to extend past ref contig end by at least N bases.
Default: 1
--maxBreak Tolerate max N unaligned bases at contig ends.
Default: 50
--motifs If set keep only reads containing given motif/s from a comma delimited list
of strings. By default also search for reverse complement of motifs.
i.e. TTAGGG,TTAAGGG will also match CCCTAA,CCCTTAA
Default: None
--noRev If set do NOT search for reverse complement of specified motifs.
Default: Find motifs on both strands.
--noPoly If set collapse homopolymer tracks within motifs before searching overhangs.
i.e. "TTAGGGTTAGGGTTAGGGTTAGGGTTAGGG" -> "TAGTAGTAGTAGTAG".
Useful for PacBio or ONP long reads homopolymer length errors. Defaut: Off.
--matchAny If set motif match may occur in unclipped region of alignment.
Defaut: False
--version Show program's version number and exit.
Teloclip-extract Options
Run teloclip-extract --help to view the programs' most commonly used options:
Usage: teloclip-extract [-h] --refIdx REFIDX [--prefix PREFIX]
[--extractReads] [--extractDir EXTRACTDIR]
[--minClip MINCLIP] [--maxBreak MAXBREAK] [--version]
[samfile]
positional arguments:
samfile If not set, will read sam from stdin.
optional arguments:
-h, --help Show this help message and exit
--refIdx Path to fai index for reference fasta. Index fasta
using `samtools faidx FASTA`
--prefix Use this prefix for output files. Default: None.
--extractReads If set, write overhang reads to fasta by contig.
--extractDir
Write extracted reads to this directory. Default: cwd.
--minClip Require clip to extend past ref contig end by at least
N bases.
--maxBreak Tolerate max N unaligned bases at contig ends.
--version Show program's version number and exit
Citing Teloclip
If you use Teloclip in your work please cite this git repo directly and note the release version you used.
Publications using Teloclip
van Westerhoven, A., Mehrabi, R., Talebi, R., Steentjes, M., Corcolon, B., Chong, P., Kema, G. and Seidl, M.F., 2023. A chromosome-level genome assembly of Zasmidium syzygii isolated from banana leaves. bioRxiv, pp.2023-08.
Yang, H.P., Wenzel, M., Hauser, D.A., Nelson, J.M., Xu, X., Eliáš, M. and Li, F.W., 2021. Monodopsis and Vischeria genomes shed new light on the biology of eustigmatophyte algae. Genome biology and evolution, 13(11), p.evab233.
Issues
Submit feedback to the Issue Tracker
License
Software provided under MIT license.


Download files
Download the file for your platform. If you're not sure which to choose, learn more about installing packages.
Source Distribution
Built Distribution
File details
Details for the file teloclip-0.1.0.tar.gz.
File metadata
- Download URL: teloclip-0.1.0.tar.gz
- Upload date:
- Size: 691.1 kB
- Tags: Source
- Uploaded using Trusted Publishing? No
- Uploaded via: python-httpx/0.27.2
File hashes
| Algorithm | Hash digest | |
|---|---|---|
| SHA256 | da2cc882b0e31db76de2cf485601ce26f28b2b95a3056d754c288d87fb9a83df |
|
| MD5 | 1fcb6e958a85099c305b62f65775b6b0 |
|
| BLAKE2b-256 | 7ff8f8f7ade4e89ada0cfe6b2b0e43e9f2ae4fc09a779308b492c0f52d509e98 |
File details
Details for the file teloclip-0.1.0-py3-none-any.whl.
File metadata
- Download URL: teloclip-0.1.0-py3-none-any.whl
- Upload date:
- Size: 19.9 kB
- Tags: Python 3
- Uploaded using Trusted Publishing? No
- Uploaded via: python-httpx/0.27.2
File hashes
| Algorithm | Hash digest | |
|---|---|---|
| SHA256 | 914d989bb909b833eeb2d131a34e8c8f3b444968e80fd9e53045668c5e56dba3 |
|
| MD5 | 25605a536c8e5a5b4b4dbd6955d5c354 |
|
| BLAKE2b-256 | 255581e760a109cb11561da2a3039175f04b75e6076011bc56f6add8f99c5e83 |







